
Frequently Asked Questions
What are you having problems with?
If you do not see what you are looking for, contact us
- Installation
- De Novo Sequencing
- Database Search
- SPIDER (Sequence Tag Homology/Mutated Peptides)
- Quantification
- Troubleshooting
The first time PEAKS is run, the licence wizard will appear automatically. There are three options:
- Activate PEAKS with a trial or purchased licence key: Pick this option if you have a licence key. Enter the key in the “Enter Licence key” pop up and PEAKS will restart.
- Register to get a free trial licence key: Pick this option if you would like to get a free 15 day trial licence key.
- Use PEAKS as a viewer: Pick this option to strictly view PEAKS projects only.
- Click on the “Activate PEAKS Manually” button in the bottom right hand corner of the licensing wizard.
- Enter the requested information. Click next.
- If you are registering a demo on a computer, you can use the process described in the “How do I Register PEAKS Studio?” question above to get a demo licence key.
- Save the licence request file and transfer it to a computer with an internet connection.
- Go to http://www.bioinfor.com/lcs20/index.jsp
- Follow the instructions on this page to request a licence key. You will receive a licence key in your email inbox.
- Transfer this key over to the computer you would like to install PEAKS on and paste it into the text box provided in the “important licence” step of the wizard. Click next, once you proceed through the licensing wizard, PEAKS should restart and be fully functional.
On this page, you will find the recommended configurations for PEAKS Studio and PEAKS Online, desktop/server solutions respectively, as well as ideal configurations.
PEAKS can. Please check our data types section for how to load data from your instrument.
The answer is yes. When you are loading data with mixed fragmentation types, select ‘Mixed CID/HCD/ETD’. PEAKS will automatically detect the fragmentation type of each scan based on the data given.
If you are using PEAKS for the first time, visit our page dedicated to supported data file formats. This will address 99% of users. For users who have updated to the latest version of PEAKS from a previous version, consider the following scenarios:
- Thermo Raw Files: In order to make reading Thermo raw file work in PEAKS, users must first uninstall the 32 bit version of Thermo MSFileReader that worked with PEAKS 6 or earlier versions. Then install the 64 bit version of Thermo MSFileReader either separately or from configure wizard in PEAKS.
- Agilent Raw Files: In order to make reading Agilent raw file work in PEAKS, the user must first uninstall the 32 bit version of Agilent MassHunter Data Access Component (MHDAC). Then install the 64 bit version either separately or run RegisterMassHunterDataAccess.bat under C:\PEAKSStudio#\MHDAC_x64 directory in Administrator privilege.
- Waters Raw Files: PEAKS supports the direct reading of Waters raw files provided Masslynx is configured.
- Local Confidence is the confidence that a particular amino acid is present in the de novo peptide at a particular position. It is presented as a percentage.
- Total Local Confidence (TLC) is the sum of the local confidence scores (0 to 1) from each amino acid in the peptide sequence.
- Average Local Confidence (ALC) is the average of the TLC. It is TLC divided by the number of amino acids in the peptide sequence.
Usually you will find good peptides at 55% and above. This does not mean that the whole sequence is the correct sequence. This is because the beginning and end of the spectrum are much more difficult to interpret when it comes to de novo sequencing due to the high mass/low mass of the ions produced by these fragments. This is why we added the colour coding to the results so you can see where in the sequence there is higher confidence.
You can definitely chop off the low scoring amino acids from the ends of the de novo sequence tags. To do this, click on the score threshold slider button (green, yellow, red symbol at the top of the de novo results). Use the slider to set a score threshold and any amino acid below that threshold will be reduced to its mass in Daltons. Once you export the tags, the last column in the excel export will follow this confidence threshold.
Bottom line: result quality. PEAKS consistently gives more accurate peptide sequences and with better confidence than other programs. This is a result of PEAKS’ global optimization algorithms and sophisticated scoring schema.
The -10lgP score is simply a p-value on an increasing scale. For example, a p-value of 1% equals a -10lgP of 20. For a given score x, its equivalent P-value is the probability that a false identification has a score greater than or equal to x. For more information on the scoring system see the following PEAKS DB Score Tutorial.
PEAKS only requires the database to be in FASTA format, and the header should contain only standard ASCII characters.
Likely the database was simply not located by PEAKS. In PEAKS, go to Configuration then Database. Make sure you have the correct FASTA database type selected in the drop down menu and the correct database path selected in the path text box.
For PTMs with a neutral loss mass, PEAKS will search for both the monoisotopic mass of the modified amino acid as well as the neutral loss mass. PEAKS does not require the neutral loss mass to be there in order for the modified amino acid to be identified. It does help if it is present.
Please refer to PEAKS DB Score Tutorial for more details.
“-1” means that a ratio was found but it was not derived from any high scoring peptides. If you want the ratio to be displayed, you can raise the peptide score threshold set in the run parameters.
The most likely reason is users must have at least two samples and the same number of fractions in each sample.
PEAKS Studio 12.5 is recommended to be installed on 64-bit Windows operating systems with Windows 10 or later.
Recommended Configurations:
Desktop License: 30+ threads processors and 64 GB+ of RAM with compatible GPU (described below)
Workstation License: 60+ threads processors and 128 GB+ of RAM with compatible GPU (described below)
e.g. Intel Core i7/i9/Xeon or AMD Ryzen 7/9/Threadripper processors
For running DeepNovo, it is required that the machine is equipped with a NVIDIA CUDA compute capability ≥ 8 GPU with at least 8GB of dedicated memory.
For running DIA Database Search, it is recommended that the machine is equipped with a NVIDIA CUDA compute capability ≥ 5 GPU with at least 8GB of dedicated memory.
The GPU must be updated to CUDA version 12.3 or later.
- Best to have all files (sample files, project, and database) stored in the local drive.
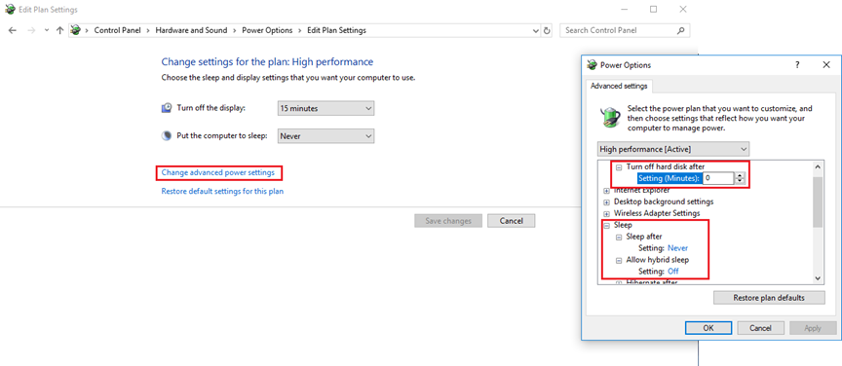
- Ensure power settings are set to never sleep
- go to the start menu and open the control panel by typing ‘control panel’
- in the top right search bar, type ‘power’ and open ‘change power settings’
- switch to the high-performance option
- also, select ‘change plan settings’ and then ‘change advanced power settings’
- find ‘turn off hard disk power’ and switch it to 0, this means the hard disk power will not turn off
- also turn all sleep options to ‘never’
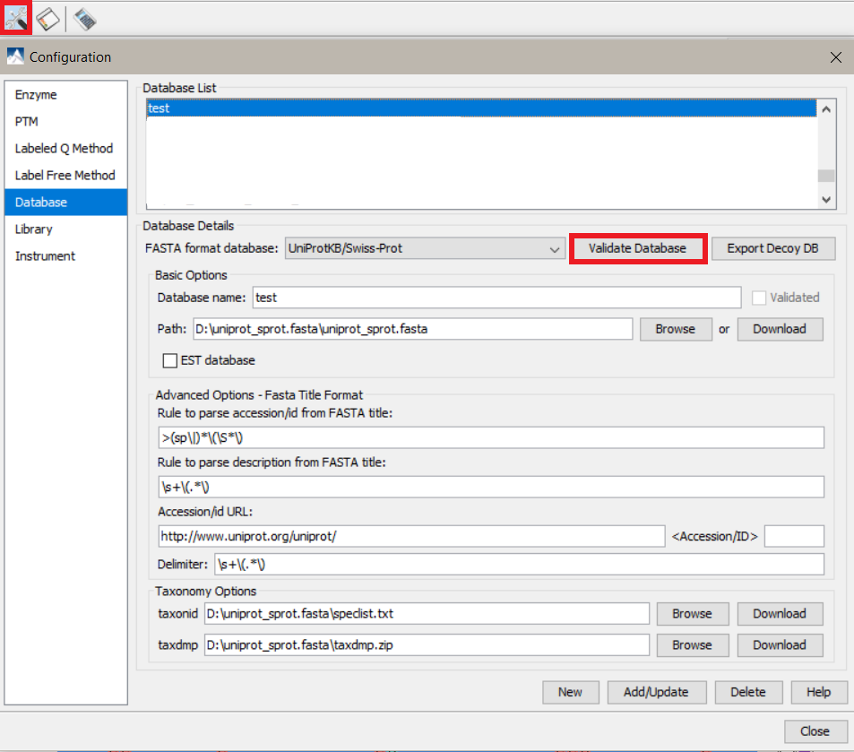
- Check if PEAKS is able to read the fasta file correctly or if there is any invalid content by going to Preferences>Database and then click the “Validate Database” option.
- Reduce batch size to prevent out of memory errors.
- go to C:\PEAKSStudioXplus\algorithmpara and open batchSizeConfig.xml in notepad
- then, change 5000 to 2500
- this will reduce the size of the batches run through PEAKS DB
- then, change 1.3 to 2.0
- this will increase the amount of memory allocated to searching each spectrum
In some cases, a PEAKS search is completed but no peptides, or very few peptides are identified. This may happen if the specified mass error tolerances are stringent for the data. Below are the suggested mass error tolerances for each instrument.
Suggested Instrument Mass Error Tolerances
| Instrument | Parent Mass Error Tolerance | Fragment Mass Error Tolerance |
| Orbi-Trap | 15.0 ppm | 0.5 Da |
| Orbi-Orbi | 15.0 ppm | 0.02 Da |
| Triple-TOF | 15.0 ppm | 0.1 Da |
| QTOF | 0.1 Da | 0.1 Da |
| TIMS-TOF | 15.0 ppm | 0.1 Da |
| TOF-TOF | 0.1 Da | 0.1 Da |
| Ion Trap | 0.5 Da | 0.5 Da |
| Linear Ion Trap | 0.5 Da | 0.5 Da |
Above are the default mass error tolerances for each instrument. These can be adjusted as necessary by referring to Figure 2b. Scatterplot of PEAKS peptide score versus precursor mass error and Figure 6. Precursor mass error of peptide-spectrum matches (PSM) in filtered result.
Other Products & Services
For questions pertaining to other products and services, please contact support.