
PEAKS Software Packages: What to select?
SOFTWARE SOLUTION FOR QUALITATIVE AND QUANTITATIVE LC-MS PROTEOMICS
PEAKS Software is a comprehensive, vendor-neutral, proteomics tool that provides systematic identification and quantification of peptides/proteins in a complex protein mixture using tandem mass spectrometry (LC-MS/MS). With PEAKS, directly load in the raw LC-MS/MS data from the mass spectrometer (no need to convert the data first) and select an appropriate PEAKS workflow to perform peptide and protein identification (such as de novo sequencing, database and spectral library searching), PTM and mutation characterisation, and quantification. The interactive interface also provides a detailed, easy-to-use, user interface for data visualisation, result validation and reporting.
Unlike other software tools, which rely solely on database searching, PEAKS uses a unique de novo-assisted database search algorithm to maximise the peptide identification efficiency for in-depth analyses of complex proteomes. By utilising this approach, you can expect to improve the accuracy and sensitivity of a traditional database search, enhance unknown modification and variant discovery, and infer new ORFs from novel peptides.
Another unique workflow in PEAKS is our DIA workflow that incorporates spectral library search, database search, and de novo sequencing in one easy to use pipeline. This workflow results in increased sensitivity and depth of proteome coverage, while keeping the search space well controlled, and thus ensures no good spectra remain unidentified.
Finally, PEAKS supports targeted proteomics approaches, such as PRM and Hybrid-PRM/DIA.
PEAKS software is available in a variety of deployment packages to accommodate various computing environments, such as desktop workstations, automated pipelines, internal servers, and cloud environments. Given its dedication to yield sensitive results while maintaining a high accuracy, PEAKS solutions can be adapted for every lab setup.
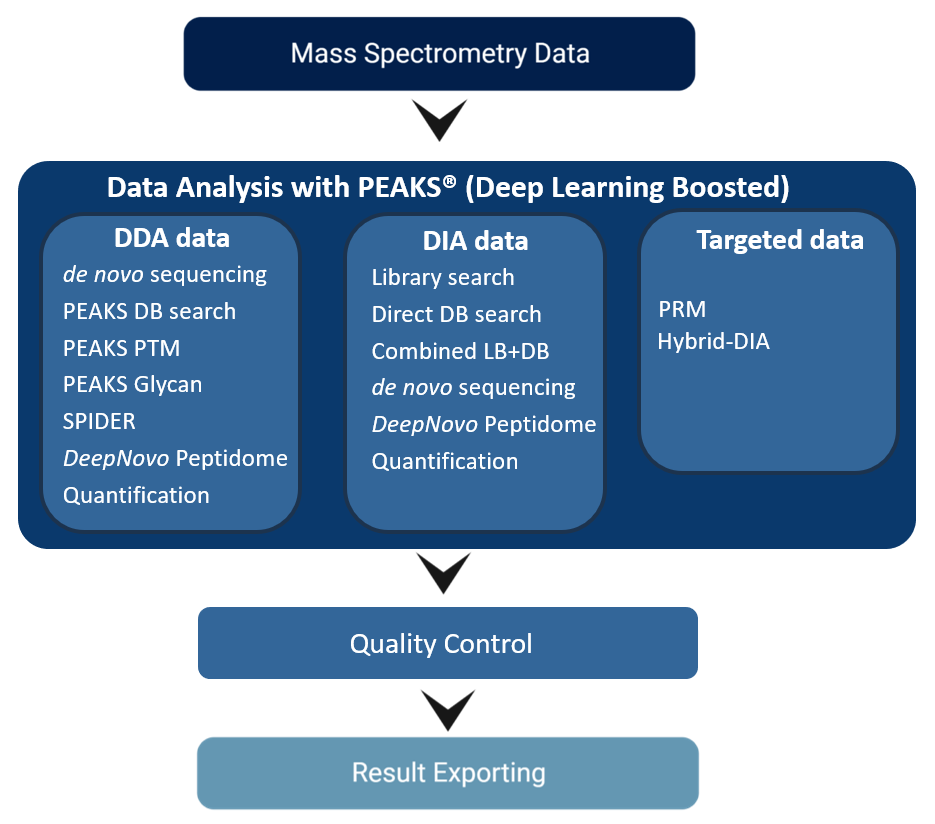
Highlights
PEAKS directly supports all native raw file formats from major vendors of mass spectrometry instruments, without any need for conversion. The required data libraries are integrated right into PEAKS to avoid any trouble or extra work with file format conversion. Working with raw data does have its advantages and by embedding these data libraries into the PEAKS algorithms the information in the data can be maximised. Regardless which instruments you have in the lab, PEAKS is finely tuned for different instrument types and is designed to ensure optimal accuracy and sensitivity for your analysis.
Both DDA and DIA technologies are rapidly advancing, and researchers need a data analysis software that combines the benefits of both acquisition methods. DDA is still a common method used in the industry and this well-established approach is especially important for test specimens that have not been well-characterised. In recent years, DIA has become increasingly popular due to its parallel nature of acquiring all fragment ions for all precursors within a selected m/z range. This overcomes the limitations of sequential MS/MS acquisition in DDA.
New DIA Workflow
PEAKS® offers a robust solution for DIA data analysis. It incorporates three methods of peptide identification: spectral library search, direct database search, and de novo sequencing. The search is performed using an expanding search space. First, a library search is performed against a library of previously identified spectra. By predicting the false discovery rate, peptides that pass the filter are saved. MS/MS spectra that don’t match a peptide within the false discovery rate threshold are brought forward to a direct database search. Confident database matches are added to the result. Then, using the same FDR approach, unmatched spectra from the database search are analysed using de novo sequencing. Learn more
Note: This option is currently only available in PEAKS Studio & PEAKS Online.
For DIA analysis, PEAKS 12.5 provides an identification workflow that allows users to analyse their data using a spectral library. With a comprehensive library, users can easily screen their data to accelerate the analysis.
Note: This option is currently only available in PEAKS Studio & PEAKS Online.
PEAKS Library Viewer
The PEAKS Library Viewer provides users the ability to visually inspect the details of their spectral library and easily assess the quality using the statistical data provided. This allows scientists to easily customise, validate, and review libraries to improve search identifications. The intuitive user interface allows you to upload and view PEAKS text format libraries generated by PEAKS Studio and PEAKS Online, as well as libraries from MSFragger, OpenMS, and Spectronaut. The third-party libraries listed can be configured into Studio from the Library Viewer.
Note: This option is currently only available in PEAKS Studio, however PEAKS Online users can still use this function by downloading PEAKS Studio and using the free PEAKS Viewer.
PEAKS is well-known for its superior result visualisation. Users have access to several different views of their results that allow viewing on a protein, peptide, and even amino acid level. PEAKS’ protein coverage view allows users to find a peptide of interest as well as pull up the associated spectra. Workflows allows even a beginner of PEAKS to easily analyse the data, while the built-in result validation guards the result quality and provides additional confidence. PEAKS graphical user interface is designed for easy interpretation, visualisation and validation to help researchers reach the overall goal of their projects.
PEAKS has become the industry standard for automated de novo sequencing, and is well known for its accuracy, speed, and ease of use. PEAKS uses a comprehensive scoring system to provide accurate de novo peptide sequencing results. Unique to PEAKS is the Local Confidence (LC) score, which is defined as the likelihood of each amino acid assignment in a resultant peptide. Learn more
PEAKS offers a unique approach to its database search workflow by combining the derived de novo sequence with the corresponding database spectrum match. De novo peptide sequences are aligned with protein database entries to provide additional information about PTMs, mutations, homologous peptides, and novel peptides. PEAKS DB uses a tag-based search algorithm, which enables search accuracy and sensitivity. With PEAKS DB, researchers can fully characterise their sample by analysing peptides whether in the database or not. Learn more
Activate Deep learning-boost in PEAKS DDA workflows to maximise peptide ID efficiency for PEAKS DB search results. Deep learning-boost improves identification accuracy and sensitivity by using algorithms to predict retention time, fragment ion intensity, mass-to-charge ratios, and ion mobility.
The identification of PTMs is achieved by integrating PEAKS DB and de novo sequencing results. The advanced algorithm included in PEAKS maximises PTM identification and PTM profiling. The identification of PTMS can be determined in any of the identification searches (de novo, PEAKS DB, spectral library search, PEAKS PTM and SPIDER). However, only in the PEAKS PTM search can users expand the PTM analysis to uncover any unexpected PTMs in their data by specifying a set list of PTMs of interest or search all 313 naturally occurring biological modifications from the Unimod database. PEAKS PTM is specifically designed to discover unexpected modifications by integrating the powerful de novo sequencing algorithm and database searching. Don’t let your computational resources limit you. With PEAKS, maximise ID efficiency and thoroughly characterise PTMs in a complex proteome.
To handle mutations, PEAKS software includes SPIDER, an algorithm specially designed to detect peptide mutations and perform cross-species homology search. The SPIDER algorithm tries to match the de novo sequence tags with the database proteins. When a significant similarity is found, the algorithm tries to use both de novo sequencing errors and homology peptide mutations to explain the differences. SPIDER aims to reconstruct a “real” sequence to minimise the sum of de novo errors between the real sequence and the de novo sequence, and homology peptide mutations between the real sequence and the database sequence. Find confident hits that do not exist in the database with PEAKS’ de novo-based homology search. Learn More
This newly developed solution is a specialised workflow for peptidomics data that combines canonical and non-canonical database searching, DeepNovo sequencing, and identification of mutated peptides. By training deep learning model using peptidomics datasets, the sensitivity and accuracy of peptide identification can significantly be improved. Furthermore, DeepNovo peptides are combined with database peptide for more accurate estimation of false discovery rate. The final output of peptides is categorised as Canonical Database, Non-Canonical Database, Homologs (mutated peptides) or DeepNovo peptides, and it can be directly exported for binding affinity and immunogenicity predictions. Learn more
Note: This option is currently only available in PEAKS Studio and PEAKS Online.
To understand the functions of individual proteins in complex biological systems, it is often necessary to measure changes in protein abundance. As an optional add-on module, PEAKS Q allows scientists to determine relative protein abundance changes across a set of samples simultaneously by labelled or label-free quantification using LC-MS/MS. Learn more
Note: This option is currently only available in PEAKS Studio and PEAKS Online.
Ion Mobility Spectrometry – Mass Spectrometry (IMS-MS) provides a compelling analytical workflow for complex biological and chemical mixtures by adding a dimension of ion separation; the 4th-dimension. With IMS-MS, ions are separated based on their mobility through a buffer gas, which provides the capability to differentiate isobaric ions based on their shape, charge, and mass mobilities. Thus, it is possible to resolve ions that may be indistinguishable by traditional mass spectrometry.
With PEAKS IMS, analyse IMS-MS data using any of the PEAKS workflows, including: de novo sequencing, PEAKS DB, spectral library search, PEAKS PTM, SPIDER, and labelled/label-free quantification (requires PEAKS Q module). The additional dimension enables increased identification sensitivity with smaller sample amounts. Easy-to-use PEAKS graphical user interface categorises the raw data into IM-MS, IM-MS/MS, and LC-IM/MS. Researchers can easily view 4-D feature detection and feature separation based on ion-mobility. Finally, ion mobility CCS or CV values are used by the prediction model. Learn more
PEAKS Glycan is an add-on module for PEAKS Studio that provides a unique solution for in depth Glycoproteomic analysis. PEAKS Glycan module utilises a glycopeptide based approach to profile the glycoproteins in your sample using LC-MS/MS data. PEAKS Glycan module presents a refined database search and newly developed algorithms to facilitate the identification and characterisation of both N- and O-linked glycans. Glycan profiling is done at specific sites within a protein (positional profiling) and compares glycopeptide abundances across samples by label-free quantification. Learn more
Note: This option is currently only available in PEAKS Studio.
Selecting a Package
| Features |  PEAKS Studio |  PEAKS CMD |  PEAKS Online |
|---|---|---|---|
| DATA TYPE | |||
| CID/CAD/HCD/ETD/ECD/EThCD/EAD | ✔ | ✔ | ✔ |
| Mixed (Alternate & Decision tree) | ✔ | ✔ | |
| DDA | ✔ | ✔ | ✔ |
| DIA | ✔ | ✔ | ✔ |
| IMS/MS | ✔ | ✔ | ✔ |
| PRM | ✔ | ||
| WORKFLOWS/ALGORITHMS | |||
| Manual De Novo | ✔ | ||
| Auto De Novo | ✔ | ✔ | ✔ |
| Database | ✔ | ✔ | ✔ |
| DIA workflow (Spectral Library Search + directDB + de novo sequencing) | ✔ | ✔ | |
| DIA DeepNovo Peptidome | ✔ | ✔ | |
| PEAKS PTM | ✔ | ✔ | ✔ |
| SPIDER | ✔ | ✔ | ✔ |
| DDA DeepNovo Peptidome | ✔ | ✔ | |
| Label-Free Quantification | ✔ | ✔ | |
| Label Quantification | ✔ | ✔ | |
| ORCHESTRATION & WORKFLOW MANAGEMENT | |||
| Established standardised workflows/search parameters | ✔ | ✔ | ✔ |
| Role based security for functionality | ✔ | ||
| Supports automated workflow execution: Allows execution of a multi-step data analysis protocol without user intervention between each step | ✔ | ✔ | |
| Manage databases (Add/Remove/Configure) | ✔ | ✔ | ✔ |
| DATA REPORTING/EXPORTING | |||
| Data/results can be exported to text file formats (.csv, .fasta, .tsv) | ✔ | ✔ | ✔ |
| Data/results can be exported to standard file format (.mzML, .mzXML, .mgf, .pepXML) | ✔ | ✔ | ✔ |
| Results can be exported to visual report | ✔ | ||
| Generate Spectral Library | ✔ | ✔ | |
| Results can be viewed using a “viewers-only” application | ✔ | ✔ | ✔ |
| TECHNICAL REQUIREMENTS | |||
| Multi-user support | ✔ | ||
| Archiving & restoring of PEAKS project | ✔ | ✔ | ✔ |
| Can be accessed from a Windows 7, 8.1, 10, or 11 Client | ✔ | ✔ | ✔ |
| Can be deployed on a Linux OS | ✔ | ✔ | |
| Support the execution of at least 2 concurrent data analysis workflows | ✔ | ||
| Can be deployed to a scalable cloud environment such as AWS | ✔ | ||
| High-throughput Processing | ✔ |