

Key Features
- Integrated Glycoproteomics and Glycomics Solutions -Identify and quantify intact glycopeptides and released glycans. New!
- In-depth Glycan profiling for both N-linked and O-linked glycan with structural confidence – Enhanced glycomics and glycoproteomics profiling with glycan structure confidence. New!
- Comprehensive Glycosylation Site Profiling – Utilise multiple enzyme digests to achieve broader and more detailed panorama profiling of glycosylation sites.
- Accurate and Flexible Quantification for both Discovery and Targeted Methodologies– Support for both label-free and labelled glycopeptide quantification to accommodate diverse experimental needs, and support PRM method for targeted quantification. New!
- Glycan de novo Sequencing – Discover novel glycans.
- Vendor-Neutral Data Compatibility – Process data from Orbitrap, timsTOF, and ZenoTOF instruments for seamless, platform-independent analysis.
- Intuitive Visualisation and Reporting – Enhanced graphical user interface (GUI) for streamlined validation, data exploration, and reporting.
Contact us to add PEAKS GlycanFinder to your research
Intact Glycopeptide Identification and Quantification for Glycoproteomics
For glycoproteomics analysis, PEAKS GlycanFinder provides two workflows for intact N-linked and O-linked glycopeptides identification and quantification from glycoproteomics data. Glycan site profiling is designed for glycosylation site determination, with result views tailored for multiple enzyme digests. Glycan sample profiling allows for label-free and reporter ion quantification at the glycoprotein, glycopeptide, and peptide level.

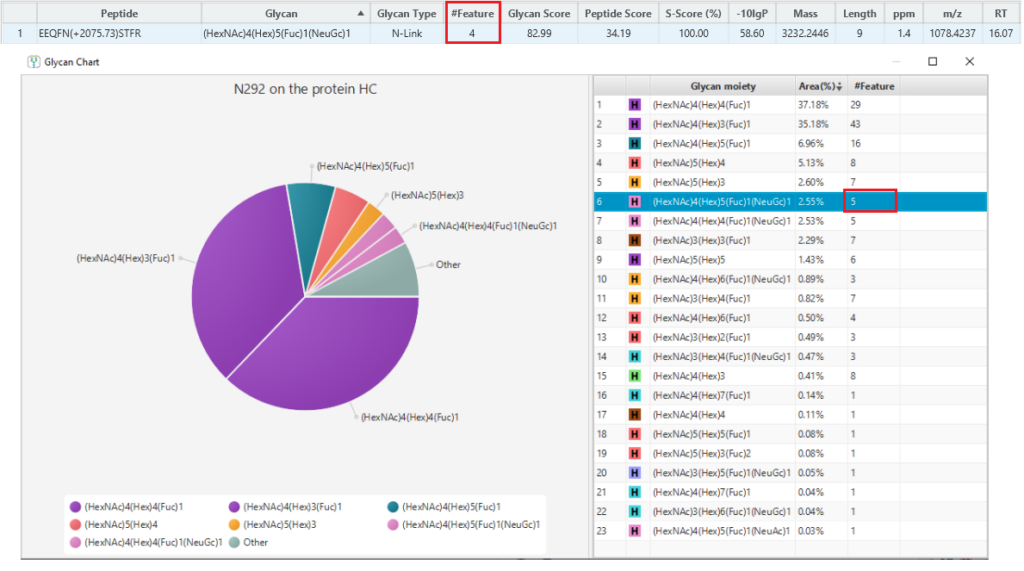
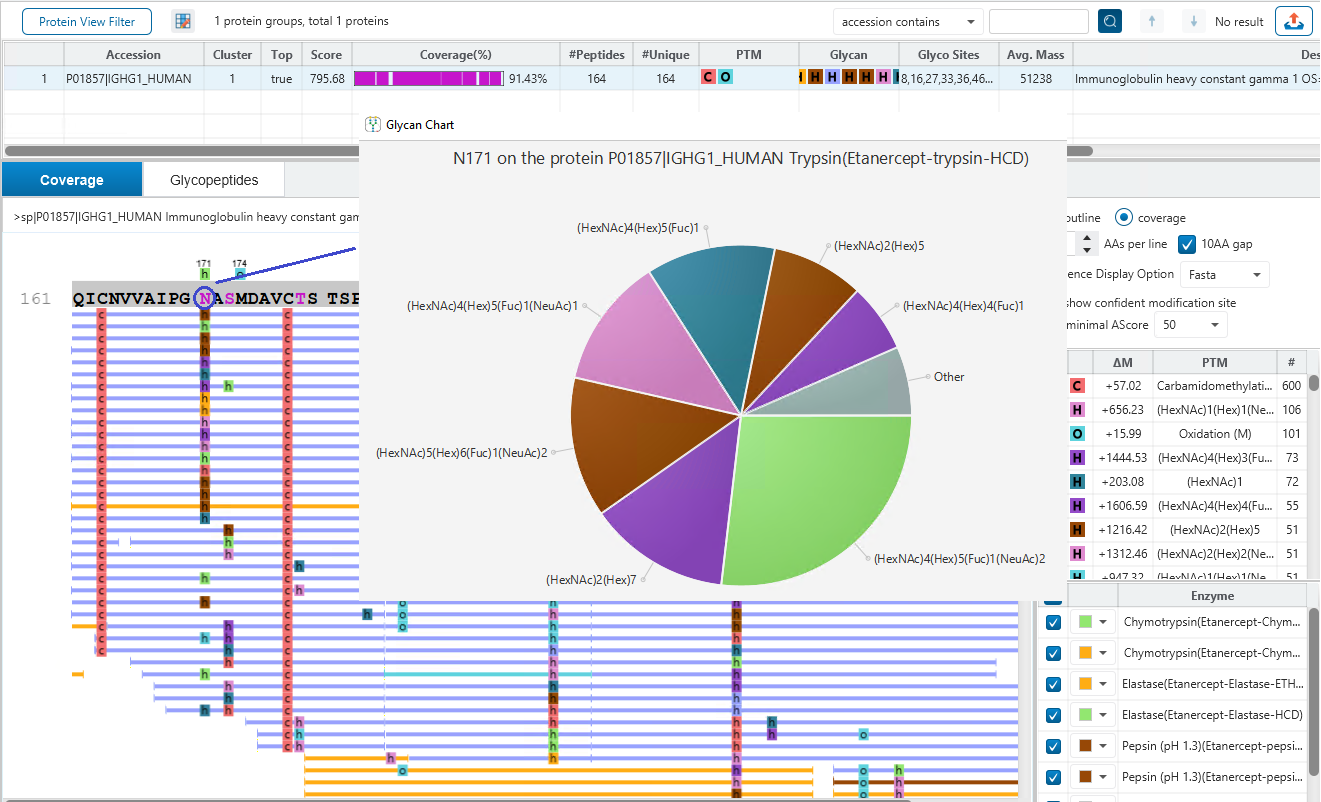
Dive into Glycan Heterogeneity – Glycan Profiling and Glycosylation Site Profiling
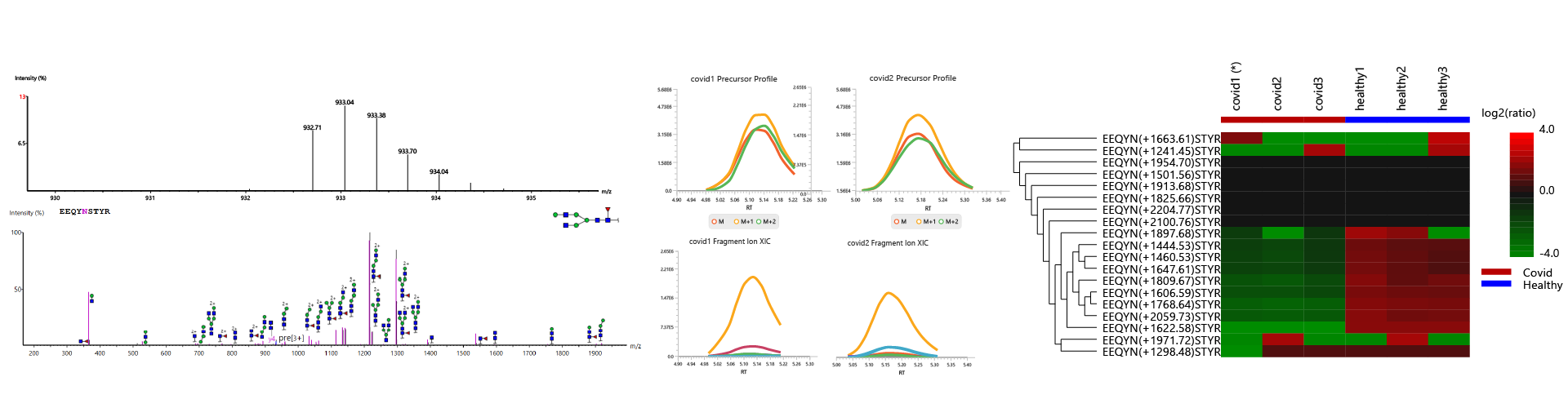
Glycan site profiling offers a detailed view of site heterogeneity, revealing the distribution and abundance of different glycans at each glycosylation site, ensuring deeper coverage of the glycoproteome. Overlay multiple enzyme digests in a PEAKS unique way to reveal the details of glycan profiling for each glycosylation site and achieve unprecedented confidence.
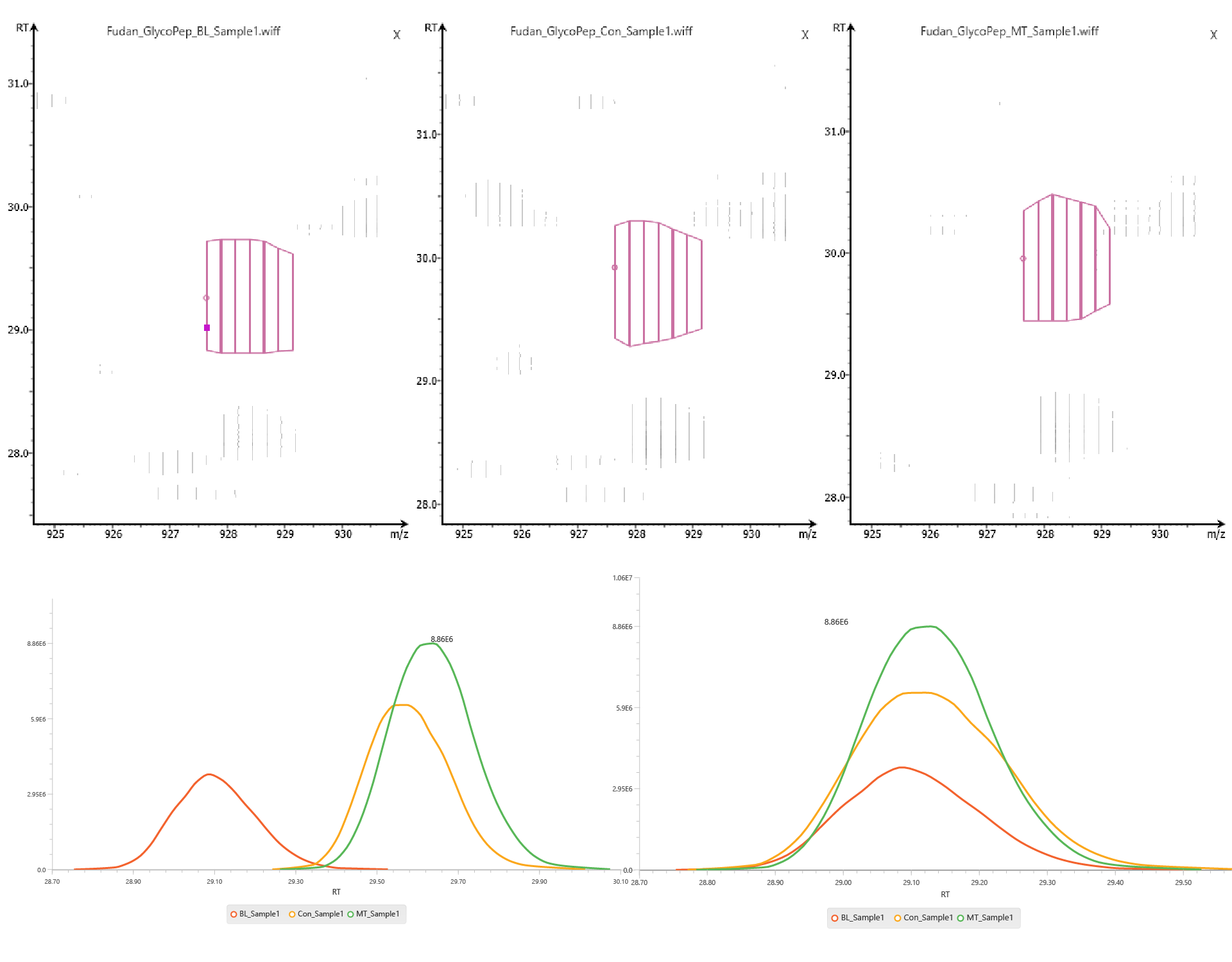
Finally, use Charge LookUp to improve the reproducibility of glycopeptide profiling, by ensuring all detected features are included even if their MS2 spectra are missing.
A peptide often produces several precursors with different charges, and not all the precursors could be identified. Normally, a peptide feature area is the sum of its all identified precursor areas. This will affect glycan profiling based on glycopeptide feature areas. To calculate the peptide feature area more accurately, Charge LookUp is applied to find the precursors of the same peptide without identification. Then, the sum of all precursor feature areas is used for glycan profiling. An example is shown in the Figure 3 below, where one feature without identification of the glycopeptide is used for glycan profiling.
Quantification of Glycopeptides and Glycoproteins
Glycan sample profiling allows for confident glycopeptide identification and direct quantification of intact glycopeptides and glycoproteins using label-free or reporter ion quantification methods.
Label Free Quantification
PEAKS GlycanFinder provides LFQ workflow to examine the relative abundance of glycoproteins. The quantification is based on the relative abundance of peptide features detected in multiple samples. Features of the same peptide from different samples are reliably aligned together via matched between runs.
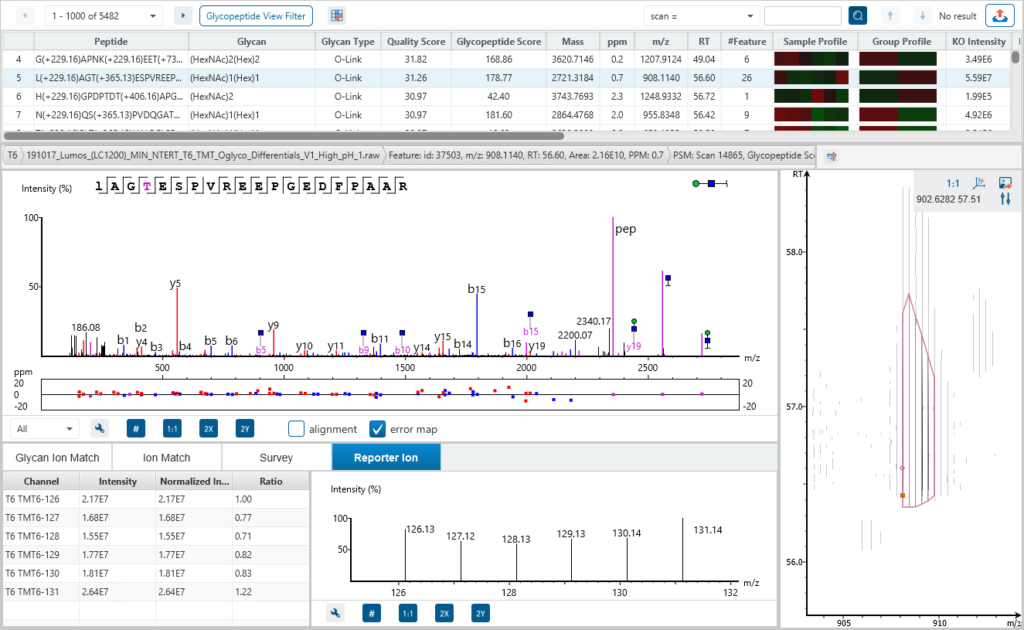
Reporter Ion Quantification
In Reporter Ion Quantification, sample ratios are derived from reporter ions of sample-specific labels. PEAKS GlycanFinder supports user-defined and commercial labels (iTRAQ, TMT).
Glycan Characterisation and Quantification with LC-MS/MS for Glycomics
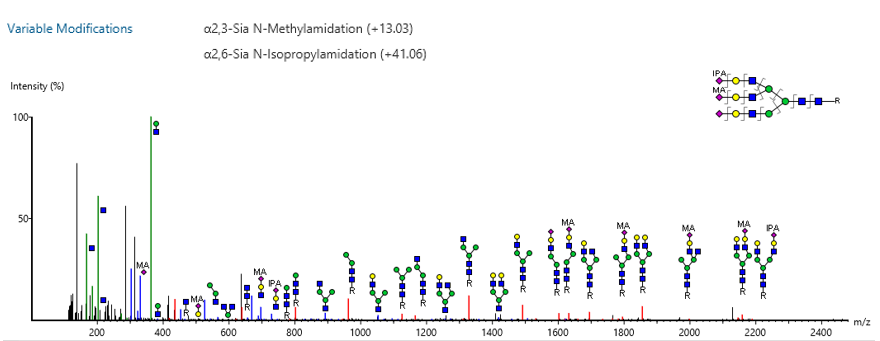
Users can bridge glycomics and glycoproteomics with the new Released Glycan Sample Profiling Workflow, which supports built-in and custom glycan modifications. Reporter ion quantification is available for released glycan quantification.
GlycanFinder now supports glycan modification and adduct searches, along with cross-ring annotation, to enhance glycan spectrum interpretation and identification accuracy.
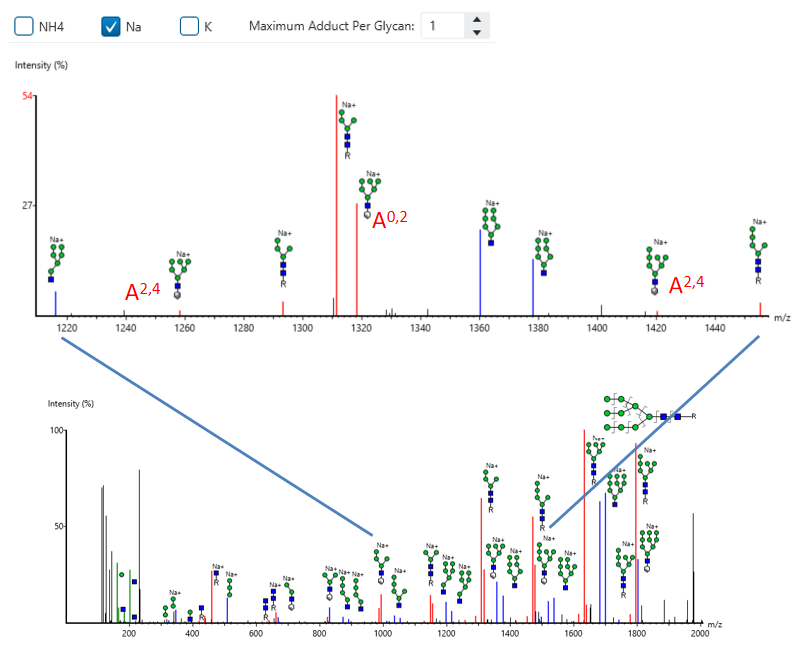
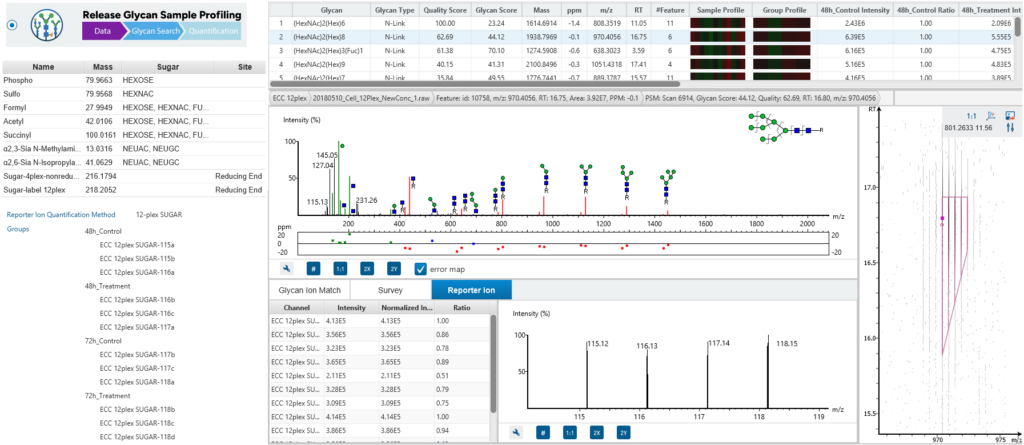
Released Glycan Reporter Ion Quantification
The released glycan RIQ enables accurate quantification of glycan abundances through the use of reporter ions. This technique improves the sensitivity and reproducibility of glycan quantification, providing more reliable data for researchers studying glycan composition and variation across samples.
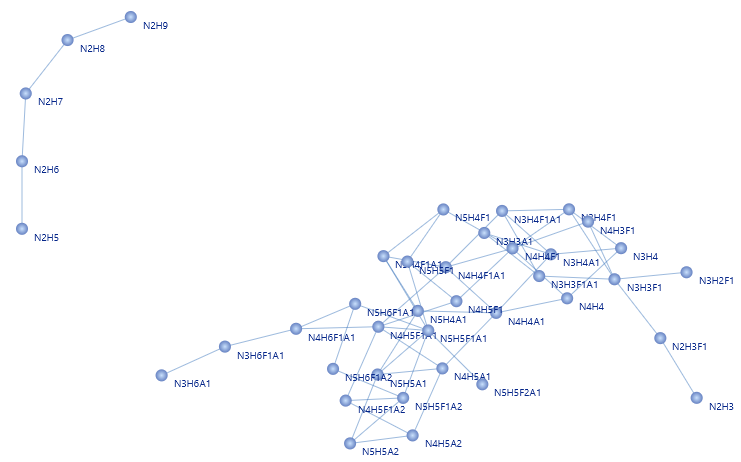
Glycan Network – Expand and Decipher Biological and Functional Glycan Associations
The glycan network illustrates the relationships between various glycan compositions, where each edge connects two glycans that differ by only one monosaccharide, making it easy to visualise the different terminal sugars extending from a common core structure. The figure of glycan network suggests that there are two clusters of glycan synthesis and processing [1].
Targeted Glycopeptide Quantification
Targeted quantification is now supported in PEAKS GlycanFinder 2.5 with the new PRM quantification workflow, PEAKS GlycanFinder quantifies and provides the MS2 structure annotation for glycopeptides in the user-defined transition list.
Unique Features
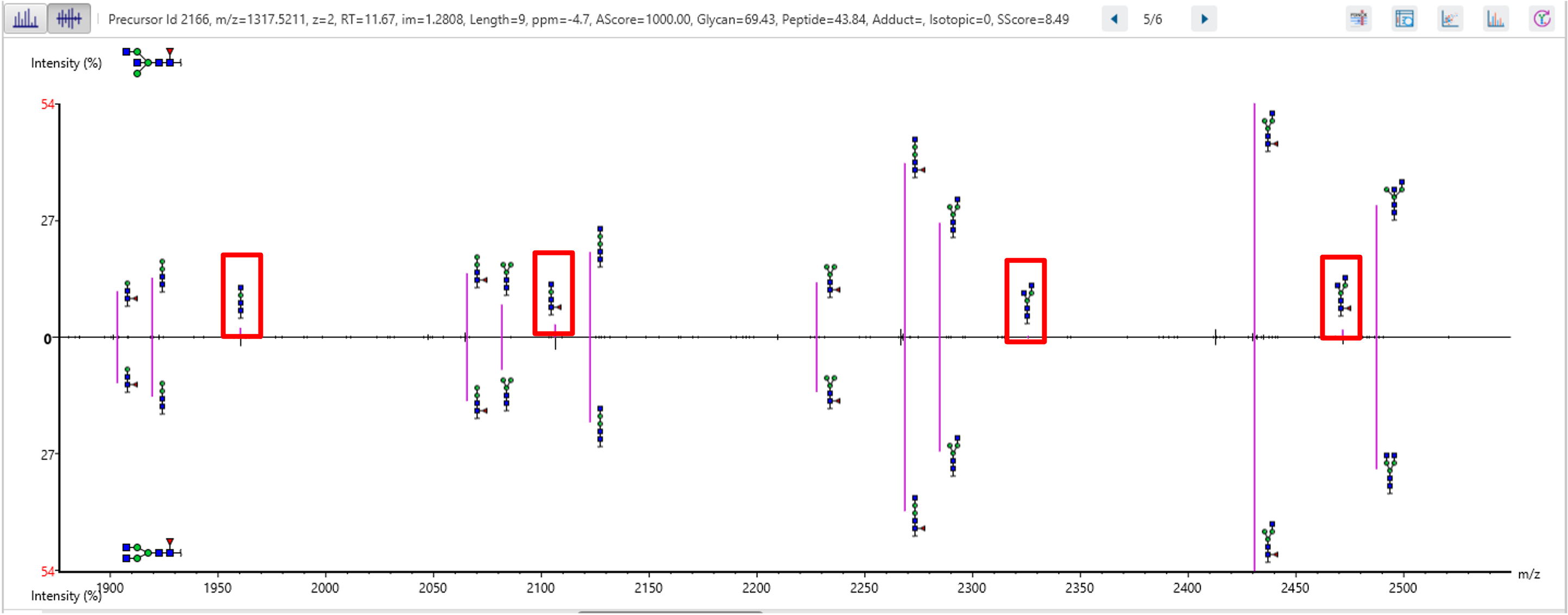
Resolve Glycan Ambiguity with Structural Resolution (S-Score)
The associated glycan on the glycopeptide is given a S-score (%), which indicates the confidence in the matched glycan structure in the glycan database.
For glycan candidates in the database with the same composition, the candidate is sorted by matched glycan Y-ion count. The higher the score the better. 100% indicates only 1 result, and it is the best match. 0% indicates that the top1 and top2 results are very similar, and we cannot confidently say the result is the best match.
Figure 11 demonstrates the use of the S-Score to distinguish between two structures with the same glycan composition. Both candidates share the same sequence and glycan moiety (HexNAc)4(Hex)3(Fuc)1. The mirror plot provides visual evidence of the fragmentation ions, helping to determine which structure corresponds to the identified glycan.
Glycan de novo Sequencing
When a glycopeptide spectrum cannot be matched to the database, PEAKS GlycanFinder will perform N-glycan de novo sequencing. This can help the user find additional glycan structures which were not considered in the database.
References
- Pinho, S., Reis, C. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15, 540–555 (2015). https://doi.org/10.1038/nrc3982
- Sun, W., Zhang, Q., Zhang, X. et al. Glycopeptide database search and de novo sequencing with PEAKS GlycanFinder enable highly sensitive glycoproteomics. Nat Commun 14, 4046 (2023). https://doi.org/10.1038/s41467-023-39699-5
- Korkola, N., Jurcic, K. PEAKS GlycanFinder 2.5: Launching a New Era for Glycoproteomics and Glycomics. Bioinformatics Solutions Inc (2025). https://www.bioinfor.com/peaks-glycanfinder-2-5-launching-a-new-era-for-glycoproteomics-and-glycomics