

New Features
PEAKS DIA Workflows
- Up to 20% increase in peptide/protein identification and quantification
- Hybrid-DIA for targeted and discovery-driven clinical proteomics (PEAKS Studio Only)
DeepNovo Peptidome workflow
- Gene table to bridge the gap between proteomics and genomics
- Support for non-canonical reference sequences
- Label-free quantification and automated QC tool
- DeepNovo-DIA peptidome workflow
DeepNovo Workflow
- Upgraded DeepNovo function with GraphNovo algorithms [1]
- GPU-enabled for increased speed
- New FDR Estimation for de novo sequencing
PRM Workflow
- New PRM Targeted Quantification (PEAKS Studio Only)
- Transition list export: from DIA LFQ results, users can export PRM and MRM transition lists
Label-free quantification
- Feature Based LFQ: LFQ result with feature vectors with and without identification.
- Label-free Quantification Principal Component Analysis (PCA) plot added to all LFQ results
- Volcano plot improvements
Post-translational modification (PTM) and Mutations analysis
- Next level confidence: confident PTM/mutation sites identification based on amino acid level evidence
User experience, automation, and more
- Batch submission option
- Database validation
- Data Refine feature filter
- Automatic export option
- Image export in SVG format (PEAKS Studio Only)
- PSM ion export


PEAKS®: Advanced Proteomics Solution
PEAKS® is a specialised tool that offers unrivalled peptide identification rates driven by deep learning technology integrated with library search, direct database search and de novo sequencing. PEAKS® 12 supports data-dependent and data-independent acquisition analyses (DDA and DIA respectively) and ion mobility mass spectrometry (IMS-MS) with the IMS add-on module. As a vendor-neutral computing platform, PEAKS® is capable of directly loading raw mass spectrometry data and standard data formats. Deploy the PEAKS® workflows to identify the presence of peptides and proteins in your project for 1) DDA data analysis including de novo sequencing, PEAKS DB (database search) identification, PEAKS PTM (post translational modification) analysis, SPIDER homology search and PEAKS DeepNovo Peptidome (specialised workflow for peptidomics data) and 2) DIA data analysis including PEAKS DIA Workflow (spectral library search, direct database searching and de novo sequencing),and DeepNovo-DIA Peptidome.
Quantification analysis by labelling and label-free quantification (LFQ) can also be performed using the PEAKS Q add-on module. Intuitive result visualisation tools are provided at every stage of analysis and results can be exported. New to PEAKS 12 users will find targeted data analysis including, PEAKS PRM Quantification, and Hybrid-PRM/DIA.
Expansive DIA solution with hyper accurate PEAKS DIA Workflow and NEW Hybrid-DIA
PEAKS® DIA workflows provide a comprehensive solution for peptide and protein identification and quantification, achieving up to 20% increase in identification rates.
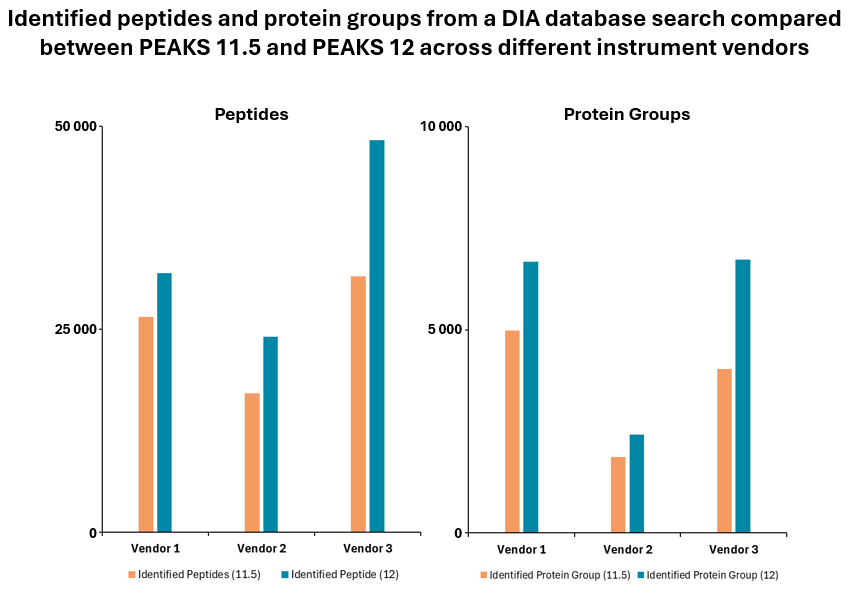
Furthermore, the platform allows for spectral library export of peptides with confident post-translational modifications (PTMs) and mutations identified in PEAKS PTM and SPIDER DDA workflows. This integration of advanced features ensures that PEAKS® DIA workflows deliver high accuracy and depth in proteomics research.
The Hybrid-DIA workflow, available exclusively in PEAKS Studio, combines targeted and discovery-driven approaches, making it ideal for clinical proteomics applications.
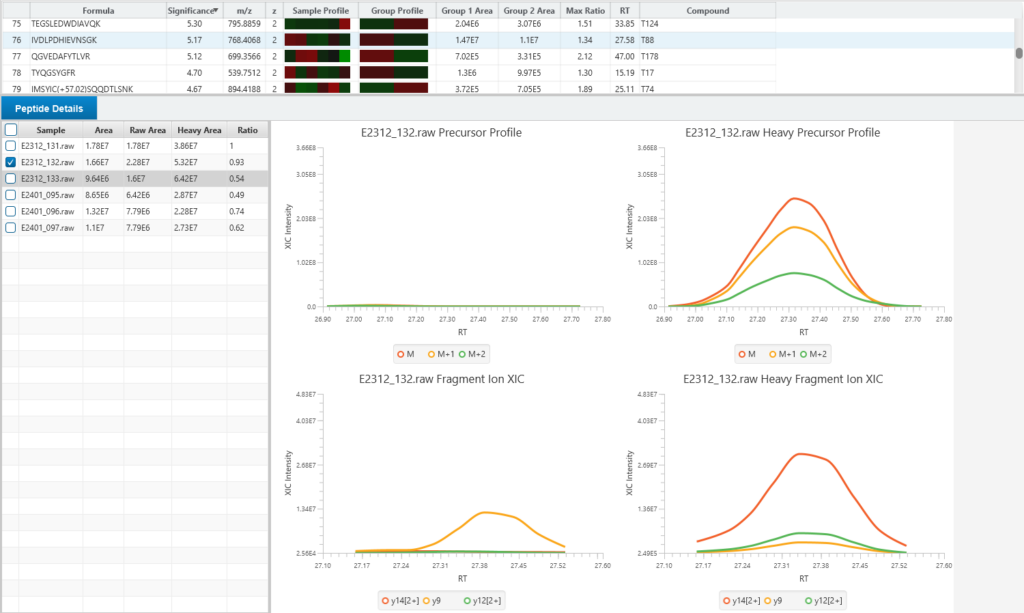
Enhanced DeepNovo Peptidome with LFQ for DDA and DIA analysis
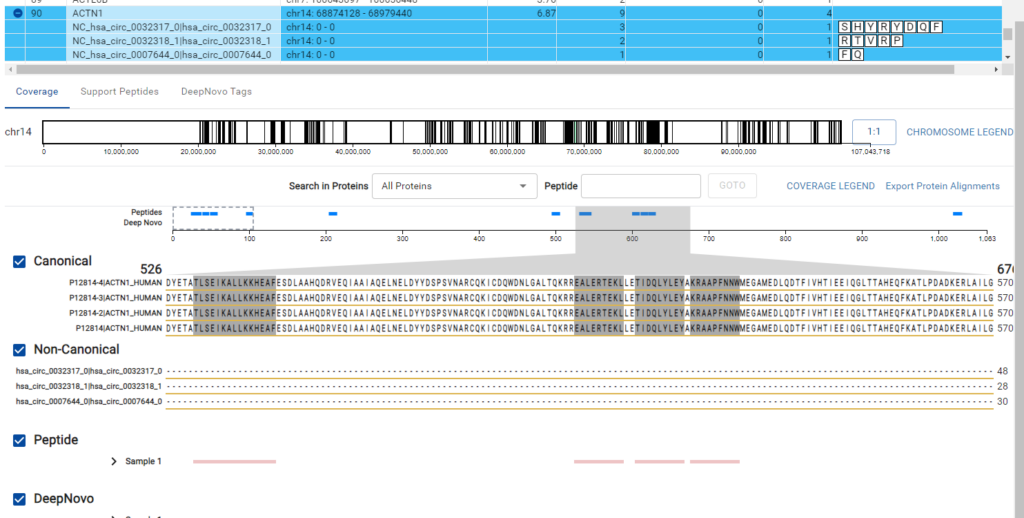
The DeepNovo Peptidome workflow is designed to bridge the gap between proteomics and genomics through the integration of a comprehensive gene table.
This feature facilitates a seamless connection between peptide identification and corresponding genomic data. The workflow supports non-canonical reference sequences, allowing for the identification of variant and modified sequences that are often missed by standard canonical reference databases.
For quantitative analysis, the workflow includes robust label-free quantification capabilities, complemented by an automated QC tool to ensure data quality and reliability.
Additionally, the DeepNovo-DIA peptidome workflow extends these capabilities to data-independent acquisition (DIA) data, providing a powerful solution for detailed peptidome profiling and ensuring high accuracy in peptide and protein identification.
The NEW upgraded DeepNovo analysis is the fastest and most accurate PEAKS peptide de novo sequencing
The DeepNovo workflow has been significantly upgraded with the incorporation of GraphNovo algorithms, enhancing the accuracy and efficiency of de novo peptide sequencing. [1]
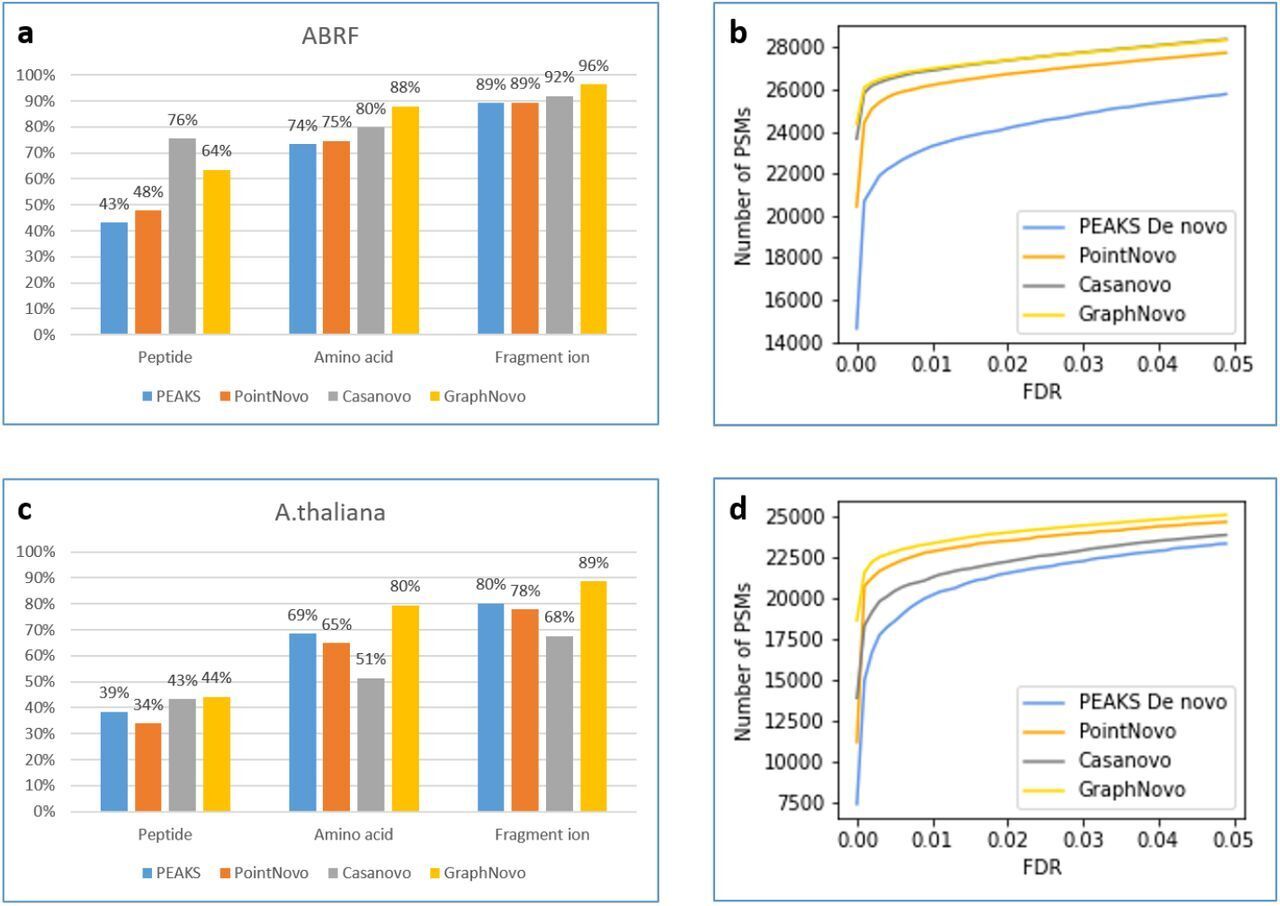
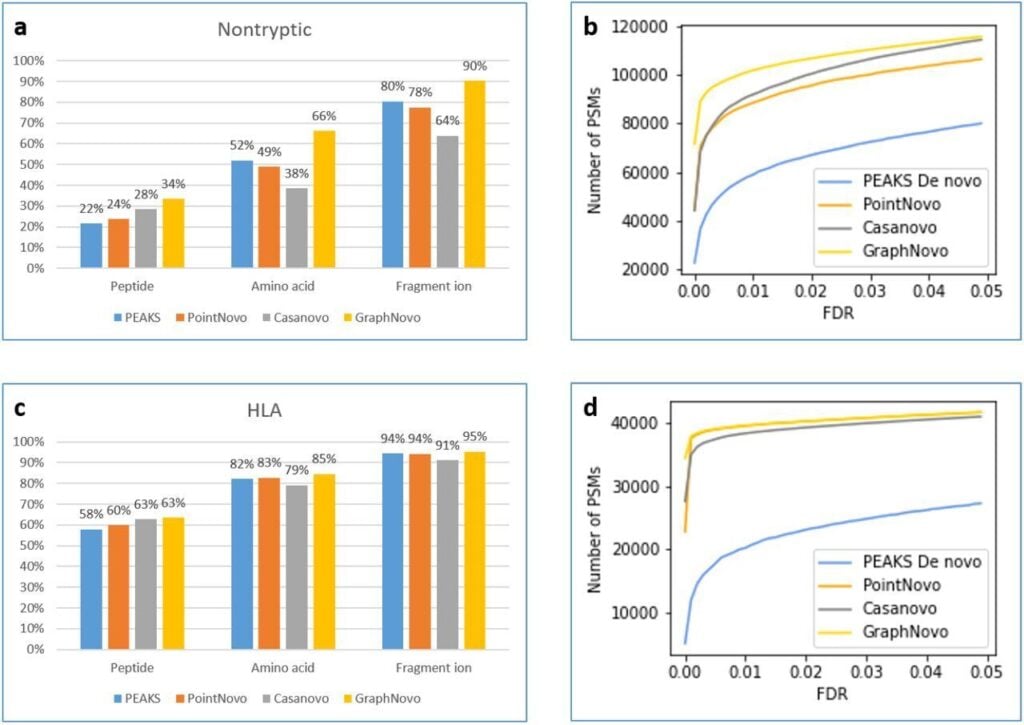
With GPU’s this upgrade leverages advanced graph-based methods to improve peptide identification, especially in complex samples. Additionally, as a GPU-enabled analysis the processing speed is dramatically increased and allows for faster data analysis
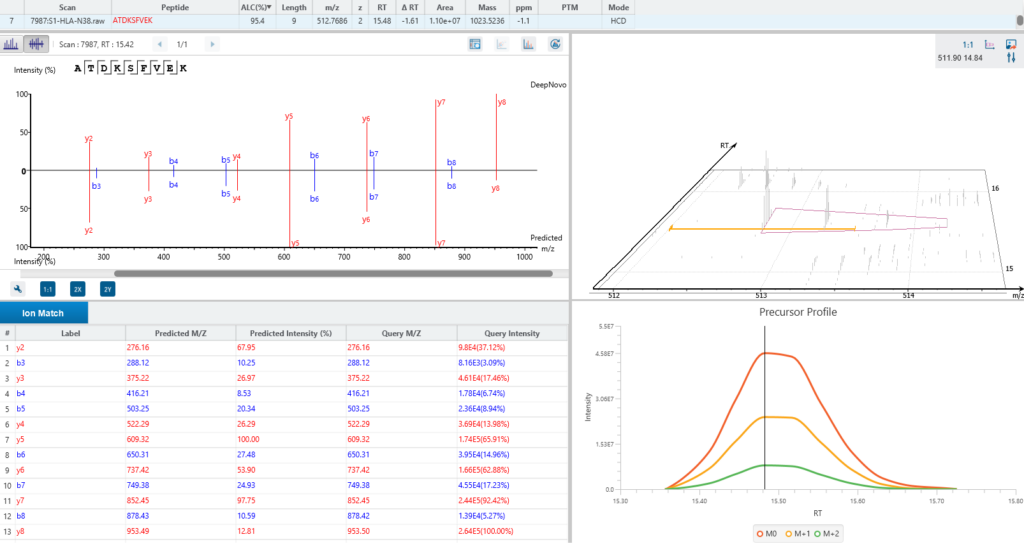
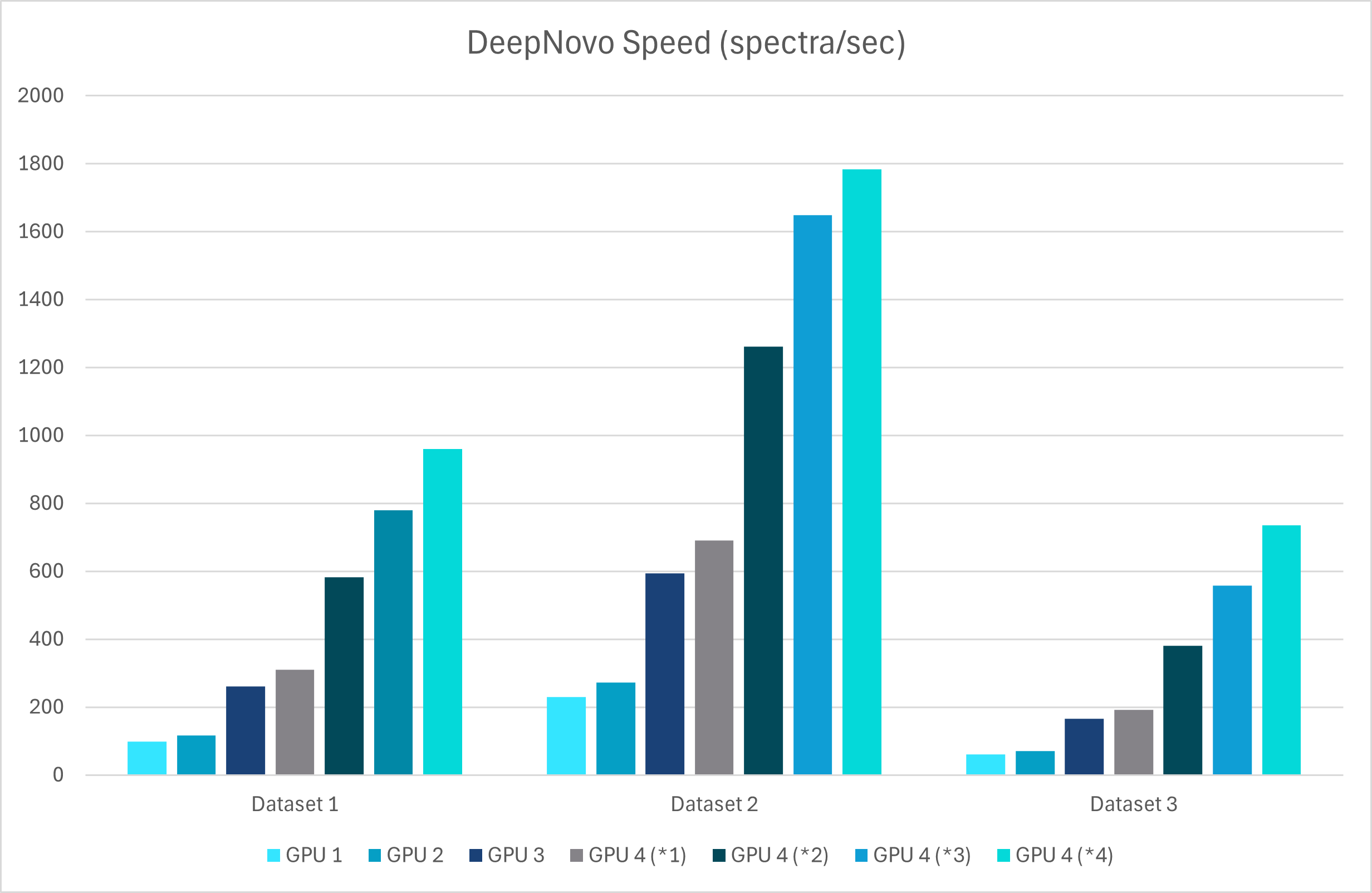
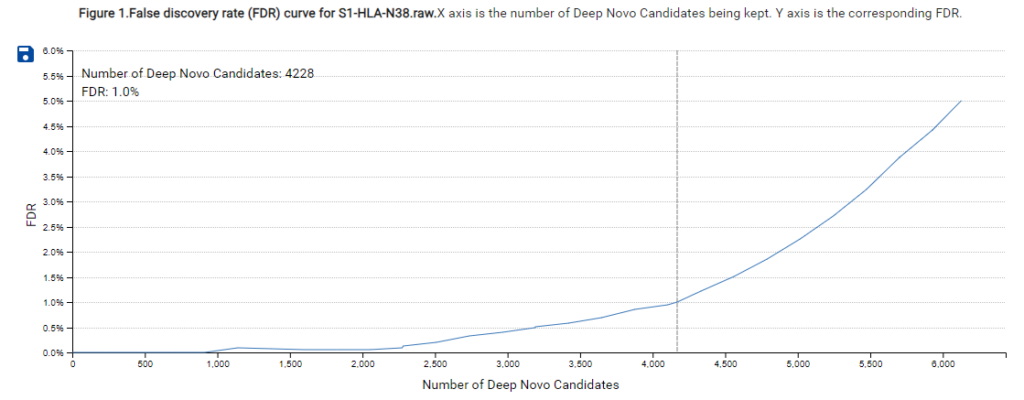
A new False Discovery Rate (FDR) estimation method has been implemented, providing more reliable and accurate control of false positives in de novo sequencing. These enhancements make the DeepNovo workflow a powerful tool for cutting-edge proteomics research, offering unprecedented speed and precision in peptide and protein identification.
PEAKS PRM is the first targeted quantification workflow integrated into PEAKS
PEAKS 12 features a new PRM Targeted Quantification workflow that is precise and reliable for quantifying specific peptides in complex biological samples.
This targeted approach allows researchers to focus on predefined peptides of interest, ensuring accurate and reproducible quantification even in the presence of interfering substances. The new PRM Targeted Quantification capability significantly improves the workflow’s sensitivity and specificity, making it an indispensable tool for detailed proteomic studies and biomarker validation. By integrating this advanced feature, the PRM Workflow offers a robust solution for high-confidence absolute quantification in targeted proteomics.
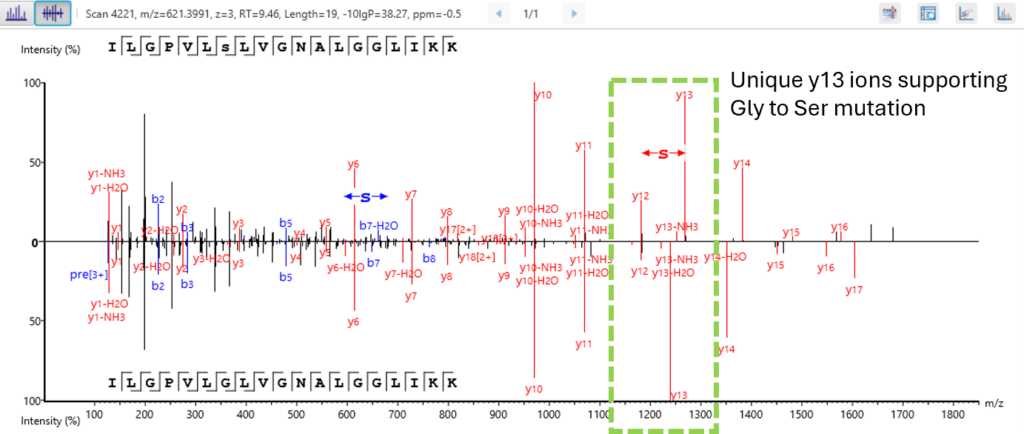
Next level confidence: confident PTM/mutation sites identification based on amino acid level evidence
The Post-Translational Modification (PTM) and Mutations Analysis workflow offers next-level confidence by enabling the identification of PTM and mutation sites with high precision, grounded on amino acid level evidence.
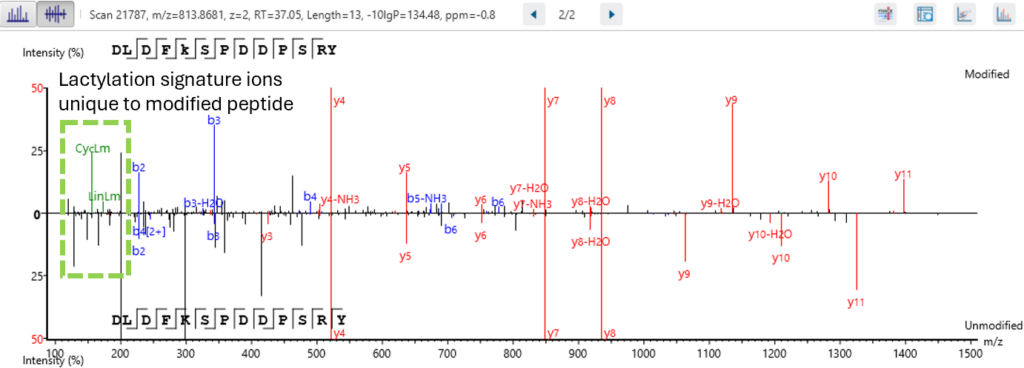
This advanced capability allows researchers to pinpoint specific modifications and mutations within proteins, providing a detailed and accurate understanding of protein function and regulation. By leveraging robust algorithms and comprehensive data analysis, this workflow ensures that PTM and mutation sites are identified with exceptional confidence, facilitating deeper insights into protein dynamics and disease mechanisms. This enhanced analytical power is crucial for advancing our understanding of biological processes and developing targeted therapeutic strategies.
References & Resources
References
- Mao, Z., Zhang, R., Xin, L. et al. Mitigating the missing-fragmentation problem in de novo peptide sequencing with a two-stage graph-based deep learning model. Nat Mach Intell 5, 1250–1260 (2023). doi:10.1038/s42256-023-00738-x