
THE NEW DISCOVERY PROTEOMICS X FACTOR

de novo sequencing | database search | spectral library search
Complete & Vendor Neutral Solution for LC-MS/MS Analysis
Highlights
- Support ion mobility mass spectrometry, including timsTOF, FAIMS, HDMSE
- Enhanced spectral library search algorithm for DIA, FAIMS DIA, SWATH, and diaPASEF
- Spectral library viewer and editor for PEAKS-generated and third-party libraries
- Advanced Label-Free and SILAC quantification with improved missing values imputation to increase accuracy and sensitivity
- Harmonised algorithms between PEAKS Studio and PEAKS Online (ex. de novo, database, spectral library, PTM, SPIDER, LFQ, TMT, and SILAC)

Advancement in proteomics technically demands sample processing, separation technique, MS instrument and data analysis. Therefore, it is important to develop software tools that are optimised to support today’s new technology and methods. Here at BSI, we aim to answer this call with our latest release in our PEAKS Studio and PEAKS Online platforms.
With advanced Ion Mobility Spectrometry algorithms and complete DDA and DIA support, PEAKS Xpro harnesses the developments in MS-based proteomics and offers a comprehensive, all-in-one solution for your lab’s LC-MS/MS data analyses. From a de novo-assisted database search to newly enhanced spectral library search, PEAKS Xpro is the new discovery proteomics X-factor!
Maximise the technology of ion mobility mass spectrometry using PEAKS Xpro with PEAKS IMS.
PEAKS IMS uses 4D feature alignment to maximise the dynamic range of identifiable and quantifiable peptides to provide in-depth analysis of complex biological samples. More importantly, this IMS information is utilised for feature detection to improve accuracy and increase precursor resolution to resolve overlapping precursor scans and chimeric spectra.
The PEAKS Xpro graphical user interface (GUI) provides many interactive data visualization tools to enhance the analysis of IMS-MS data. Through the GUI, users have the potential to resolve ions that may be indistinguishable by traditional mass spectrometry by interrogating data at the ion mobility level (ex. CV and CCS information).
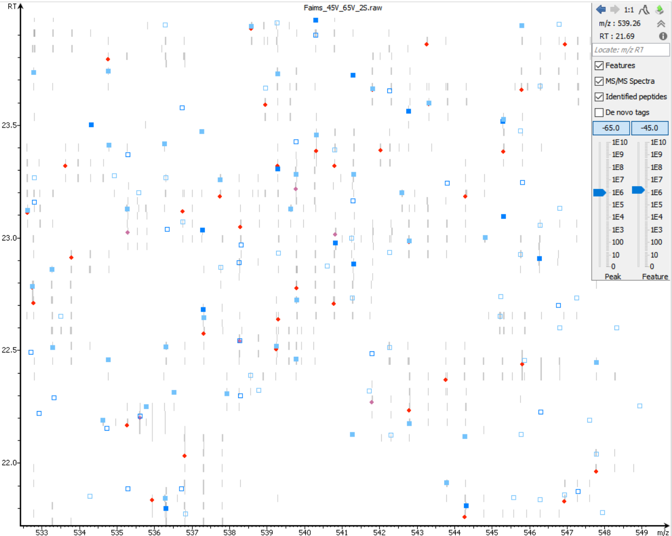
Furthermore, 4D IMS-MS information can be further utilised to increase the accuracy of quantification studies. With PEAKS Xpro, the PEAKS Q module will incorporate 4D level information to perform label-free or labelling quantification analyses. Users will also be able to conduct large-scale quantitative studies by bringing the PEAKS Xpro algorithm to the PEAKS Online platform. By completing the entire analysis in one comprehensive project, users will be able to get a full understanding of the big picture.

In the new PEAKS Xpro, users will also be able to analyse their 4D DIA data using the enhanced PEAKS spectral library search (FAIMS DIA and diaPASEF).
Generate reproducible, accurate identification of known peptides in DDA and DIA datasets with the spectral library searching in PEAKS Xpro. The enhanced PEAKS Xpro spectral library search uses m/z, intensity values, normalised retention time plus ion-mobility spectrometry values (CCS/CV values with PEAKS IMS add-on module), and is designed to work with both DDA and DIA spectral library searches. With precise FDR estimation users can detect more peptides in each dataset without sacrificing accuracy for both, DDA and DIA analyses. The addition of the spectral library search to the current functions in PEAKS allows users to perform their whole pipeline in one software tool, from creating a spectral library to spectral library searching.
Spectral libraries generated in PEAKS are comprehensive libraries. This means that the library contains both matched and unmatched peaks, the RT and IMS information, and the associated database protein. By producing comprehensive libraries, PEAKS spectral library search will take advantage of this information to improve the performance of the search. Once the library is generated, use the PEAKS Studio Xpro library viewer for quality control charts and library validation.
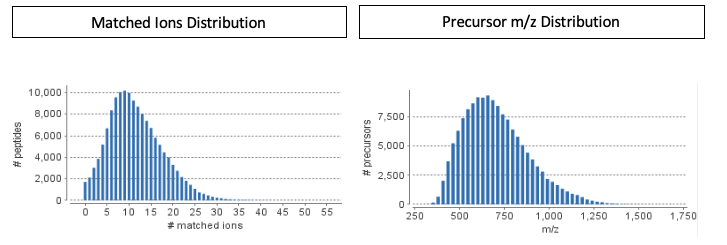
Using PEAKS Xpro spectral library search you can expect a fast, accurate and easy use solution to analyze your DIA, FAIMS DIA, SWATH, and diaPASEF. Spectral library searches are a great way to screen samples for known peptides/proteins and with PEAKS Xpro you expect; improved sensitivity for single cell proteomics; and other low abundant data. For large-scale proteomics research projects, PEAKS Online Xpro is the best solution to provide a high-throughput, scalable platform that can not only handle larger cohorts but also process them accurately and efficiently.
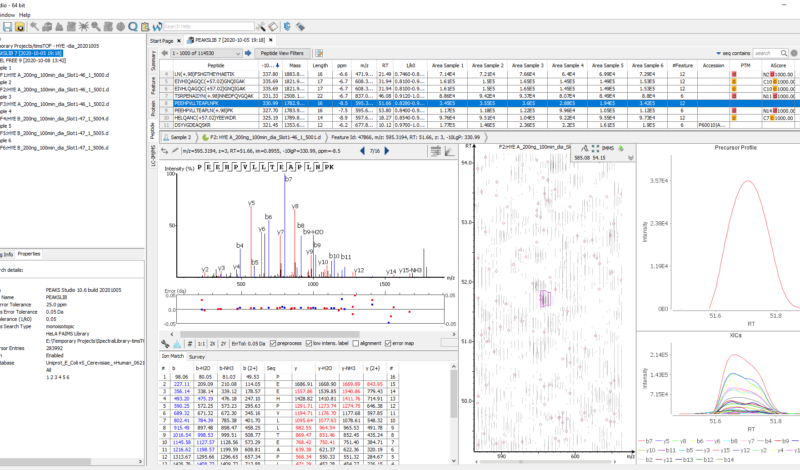
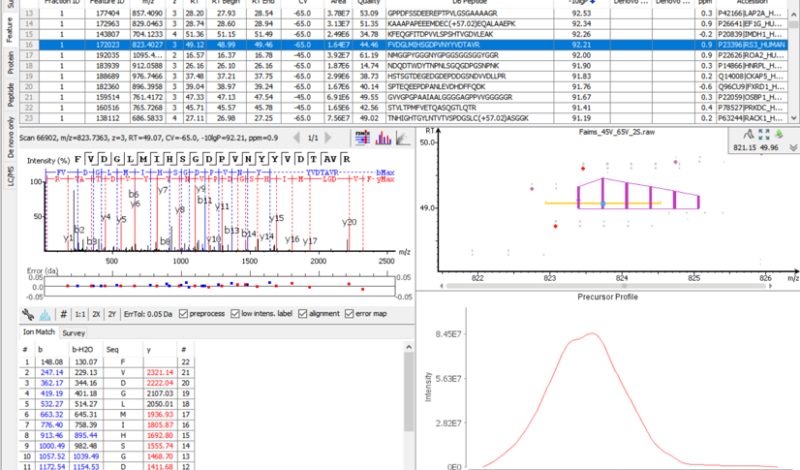
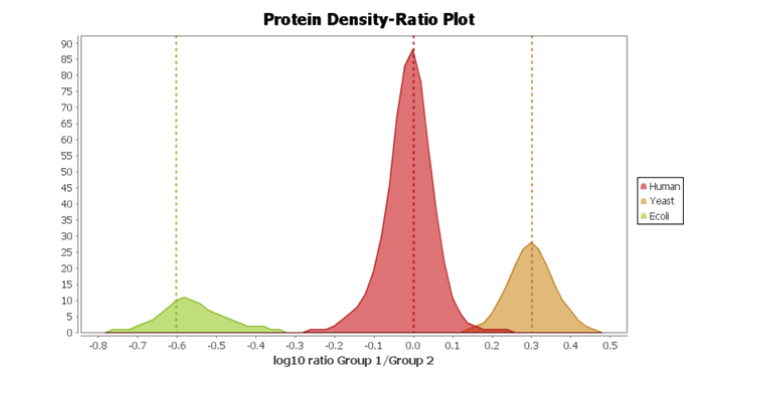
Following the new identification workflow, users can perform a quantification analysis of their data. In the example below, a SWATH dataset acquired from PXD002952 consisted of 2 samples, with 3 replicates each. The samples consisted of 3 different proteomes, which were spiked in different ratios (Human 1:1, Yeast 2:1, and E.coli 1:4). Using PEAKS Xpro, a spectral library search and then a database search was performed, followed by a label-free quantitation assessment. Using the new Density-Ratio plot provided in the LFQ result node, users can easily visualise the actual ratio between groups where the dotted line indicates the expected ratios.
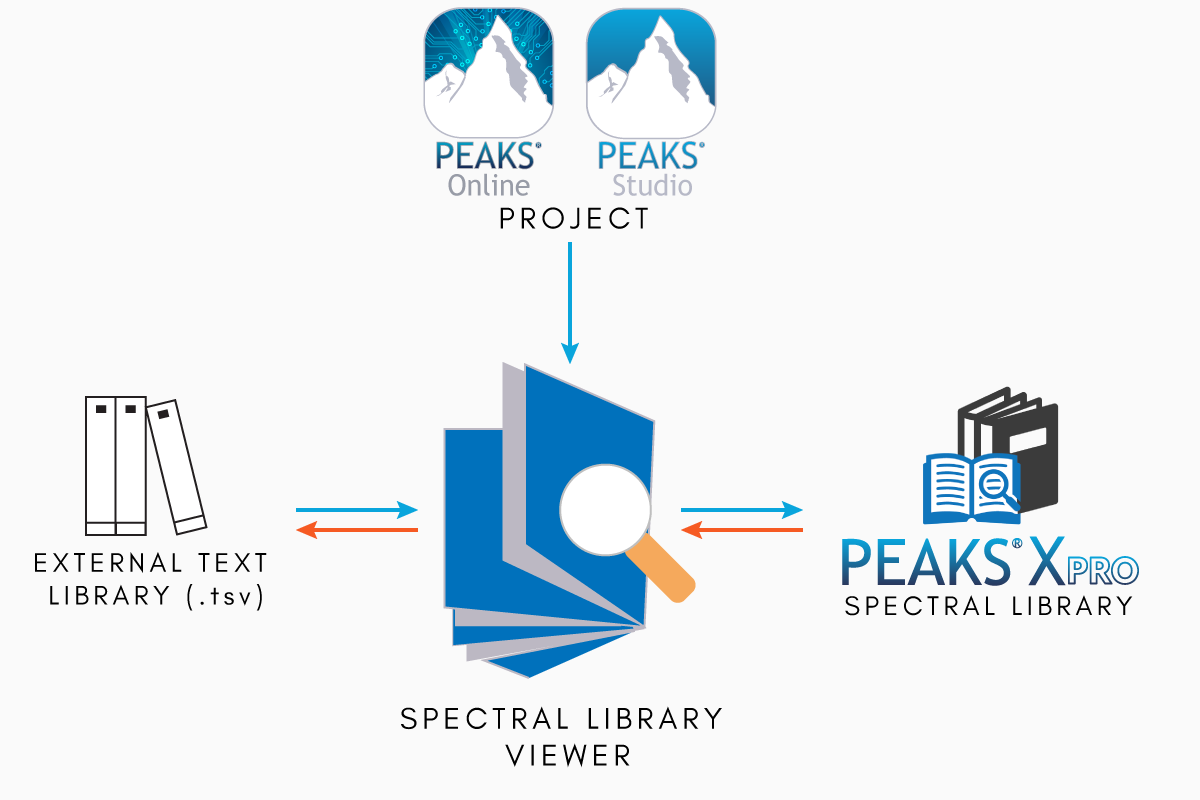
Utilise the Spectral Library Viewer and Editor within PEAKS Xpro to assess core spectral library information and gain important insight into the attributes of the library you are utilizing for your search. With detailed tables and interactive view of the spectral library, you can validate the library and easily make the necessary changes to ensure a high-quality peptide spectral library is used for all of your searches.
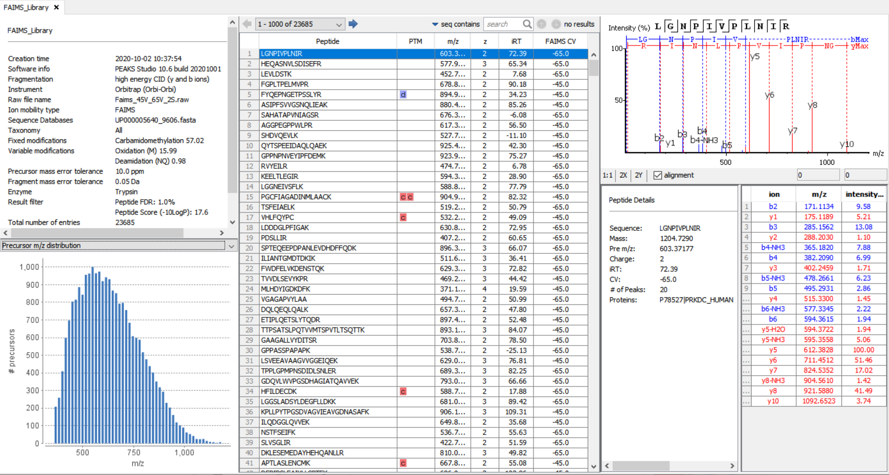
The Spectral Library Viewer is equipped with several statistical analysis of the library for quality assessment and validation. A peptide table that includes all spectral library entries and important details like peptide sequence, PTMs, m/z, z, normalised rt, ion mobility values associated with each entry. Selecting an entry in the peptide table displays an annotated peptide spectrum plus peptide details and an ion table associated with the peptide.
Spectral Library editor and viewer compatible with PEAKS_LB, OpenMS_LB, text file libraries, and more. Easily view third party libraries and utilise them in PEAKS with Spectral Library Viewer!
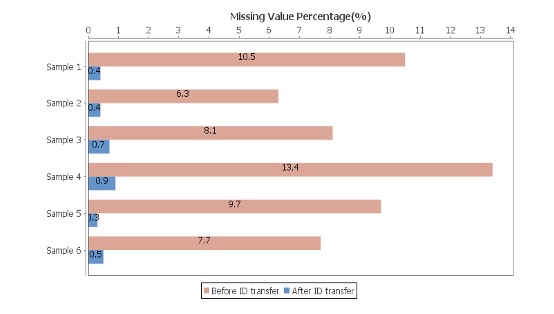
The new PEAKS Xpro features a second round of feature detection and alignment to maximise the information in quantification studies with biological and/or technical replicates. ID-transfer is used to reduce the number of false missing values which are caused by the stochasticity of data-dependent acquisition (DDA) and limited quantitative depth using spectral library-based peptide identification in data-independent acquisition (DIA).
In PEAKS Xpro, users can now see the efficiency of PEAKS ID-transfer between runs (also known as match between runs) and expect low number of missing values for LFQ and SILAC analyses.
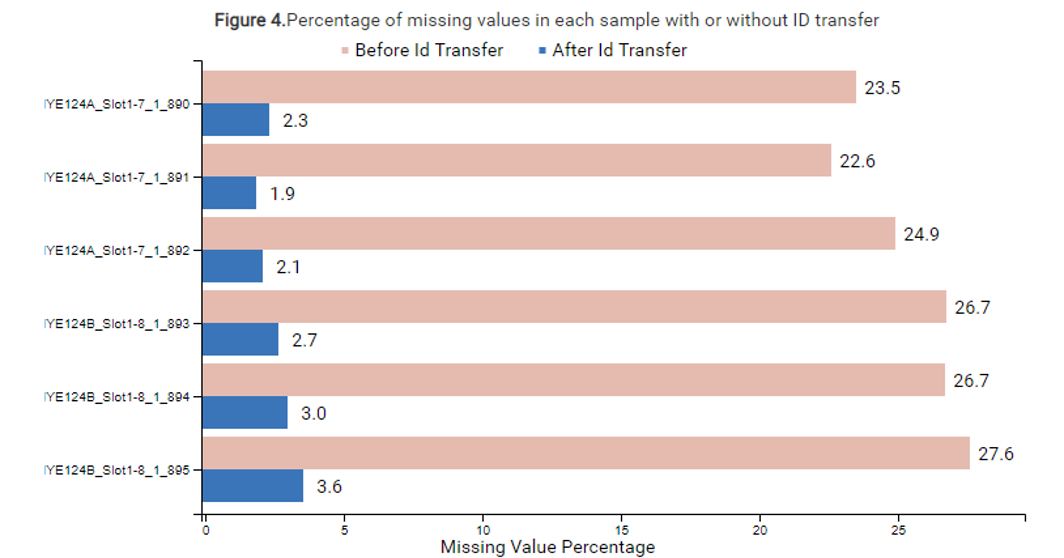
At the beginning and end of a MS run, RT tends to be highly variable and with a fixed window a large RT shift can be problematic for RT detection. To resolve this, PEAKS Xpro features an RT autodetection to improve the detection and alignment of features to provide an accurate quantitative analysis tool.
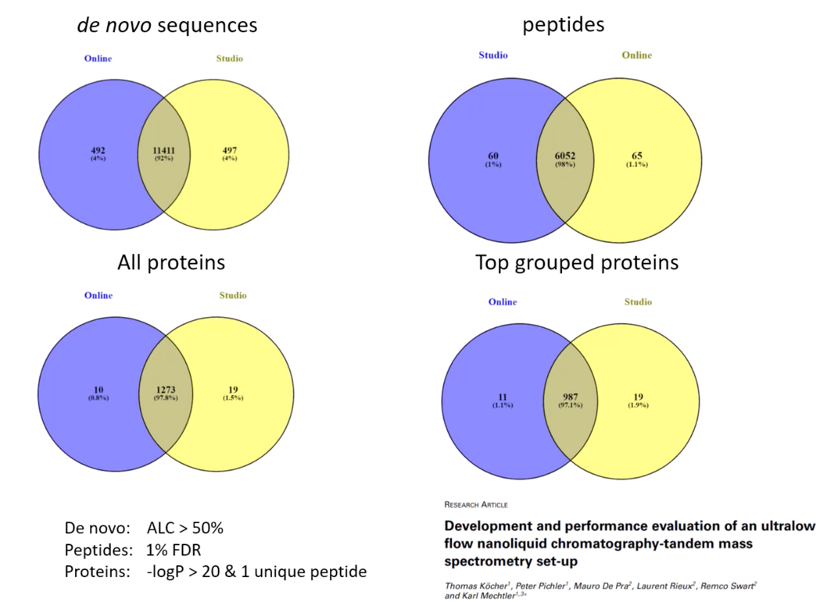
PEAKS Studio and PEAKS Online provide a similar solution and share the same trusted algorithms for de novo sequencing, database search, spectral library search, PTM, Spider, TMT, SILAC, and LFQ. With the same core functionality, you can expect a similar data analysis solution from both products and can focus on aligning the software that provides the best computation solution and user experience for your group.
PEAKS Studio Xpro is comprehensive software for detailed identification and quantification analyses. With an intuitive and easy-to-use design, PEAKS Studio provides tool to assess your data from every angle. Easily jump from spectrum to peptide to protein to validate your results. Learn more about the versatility and strength of PEAKS Studio to see how you can perform everything from de novo sequencing and spectral library search to accurate quantitative analyses.
PEAKS Online Xpro is a high-performance, multi-user solution to accelerate LC-MS/MS analyses. Designed to support large-scale proteomics studies, PEAKS Online is a high-throughput software solution that is not only easy to use and fast but also highly accurate with complete DDA and DIA support. Learn more about PEAKS Online to see how you can perform all identification and quantification analyses from standard projects to large-cohort clinical proteomics studies.