

To understand the functions of individual proteins in complex biological systems, it is often necessary to measure changes in protein abundance. For example, Biomarker discovery and validation studies typically require quantitative analysis of proteins to identify and verify potential biomarkers that show protein expression changes significantly between different states/conditions. PEAKS Q allows the scientist to determine relative protein abundance changes across a set of samples simultaneously by labelling or label-free quantification using LC-MS/MS.
Label-Free Quantification
Protein relative quantification in PEAKS is performed using ion peak intensity on MS1. In label-free quantification experiments, samples are separately collected, prepared and analyzed by LC-MS/MS. Because of the large amount of data collected from these experiments, sensitive and accurate algorithms are used in PEAKS for automated ion peak alignment and comparison. Peptide/protein identification from MS2 by database search is integrated for protein quantification.
Peptide Quantification

Same peptide ions from different LC-MS runs are aligned. Ions with different charges are merged.
Protein Quantification
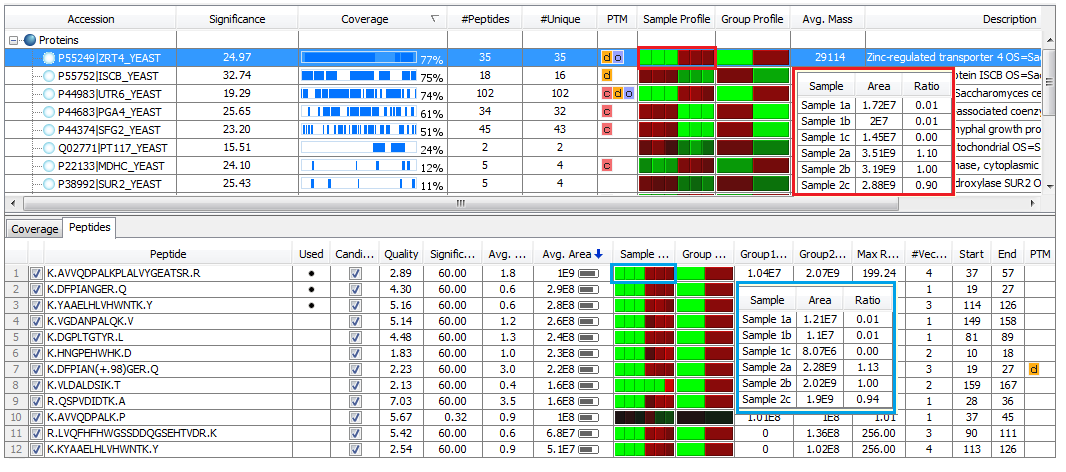
Three most abundant peptides are used for protein ratio estimation.
SILAC Quantification
SILAC has become the most common approach for in vivo isotopic labelling. Because heavy and light samples are combined before sample preparation for MS analysis, the level of quantification bias from processing errors is low.
Quantification of peptides with isotopic labelling

TMT/iTRAQ
Isobaric tags (TMT or iTRAQ) have identical masses and chemical properties that allow heavy and light isotopologues to co-elute together. The tags are then cleaved from the peptides by collision-induced dissociation (CID) during MS/MS, which is used for quantification.
One of challenges for such methods is reporter ion ratio distortion resulting from fragmentation of coisolated interfering species. The MultiNotch MS3 method address this issue by uniquely combining multiplexing capacity with quantitative sensitivity and accuracy. PEAKS supports both MS2 and MS3 quantification.
Quantification of peptides with isobaric labelling

With PEAKS, you can now expand your sample size for large-scale protein quantification studies, with reference channels to ensure the accuracy of quantification.
Data analysis for combining experiments

References
- Yang, W., et al. PEAKS Q: Software for MS-based quantification of stable isotope labeled peptides. ASMS. WP531. 30/5/2006.
- Xin, L. et al. New Quantitation Software Package Based on PEAKS Protein ID. ASMS. TP 653. 2/6/2008.
- Chen, C., et al. New Algorithm for Label-Free Protein Quantification. ASMS. MPB 043. 31/05/2009.